Ph pozitívní (Ph+)
Definice: je klonální myeloproliferativní nádorové onemocnění postihující hemopoetickú pluripotentní kmeňovú buňku. S charakteristickou cytogenetickou změnou t(9,22) Ph chromozom (filadelfský chromozom).
Epidemiologie:
- leukémia dospělých,↑muži, nad 50let
- 15% všech leukemií
- 1-2/100.000 obyvatel , ↑↑ s věkem
- 20 – 25% leukemií v dospělosti
- 3 – 5% v dětství (vzácně)
Etiopatogeneze:
Etiologie: ionizujíci záření s latenci 4 – 11let, benzen
Patogeneze: filadelfský chromozom → translokace 9/22 → přesun c-ABL (tyrozinkináza) z 9 na 22 chromozom ku BCR = vzniká fúzní gen BCR-ABL (tyrozinkináza) → ↑aktivita tyrozinkinázi, zástava maturace, autonomia proliferace, neschopnosť apoptózy
Ph chromozom → způsobuje instabilitu myeloidních buněk → ďalší mutace → nové klony → blastická transformace
Klinický obraz: 3fáze: chronická, akcelerovaná, blastický zvrat
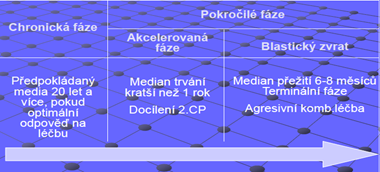
Chronická fáze: mírne, neurčité obtíže
- Krevní obraz: leukocytóza, až 30% dg. náhodně
- První příznaky: únava, bolest kloubů, svalů, subfebrilie, pocit tlaku pod levým obloukem žeberním (splenomegalie), bolest (mechanické zvětšovaním, napínaní pouzdra), noční pocení, nechutenství, hubnutí, hepatomegalie…
Akcelerovaná fáze: nádorové kmeňové buňky se stávají agresivnějšími a zvyšují rychlost proliferace, ztráci citlivost na léčbu
Blastický zvrat:
- Rozlišujeme 2 blastické zvraty: myeloidní /lymfoidní
- Krevní obraz: rozvinutá akutní leukemie
- Příznaky: teploty, infekční komplikace, bolesti v kostech, významně unavený v souvislosti s anémii, krvácení + trombocytopenie, splenomegalie
- Kostní dřeň: hyperplastická (↑granulocytů – všetky stadia, erytropoéza ↓↓)
Diagnóza:
- Anamnéza a fyzikální vyšetření:
- Fyzikální vyšetření: splenomegalie (značného rozsahu: může zasahovat až do malé pánve), hepatomegalie (mírná až střední)
- Laboratorní vyšetření:
- Krevní obraz s diferenciálem:
- Chronická fáze:
- leukocytóza (50 – 250×109/l) s posunem doleva (v diferenciálu najdeme všechny vývojové dif. řady granulocytů)
- trombocytóza (u části nemocných)
- eosinofilie + bazofilie
- anémie – mírna
- blasty málo < 2%
- Akcelerovaná fáze + blastický zvrat:
- blasty 10 – 19%
- těžká anemie
- trombocytopenie
- Aspirát kostní dřeně:
- morfologické vyšetření kostní dřeně: myelogram
- cytogenetické (klasická, FISH)
- popř. cytochemické, cytoflowmetrické (FCM)
- Trepanobiopsie (histologické vyšetření KD)
- PCR – potvrzení přítomnosti fúzního genu BCR-ABL
- Biochemické parametry včetně kyseliny močové, beta2mikroglobulinu, LDH, minerálů
- Zobrazovací metody:
- UZ břicha se změřením na slezinu, játra (staging choroby)
Diferenciální diagnóza:
- Jiná myeloproliferatívní nemoci:
- Polycythaemia vera
- Esenciální trombocytemie
- Idiopatická myelofibróza
- Reaktivní leukocytóza: infekce, hemolýza, krvácení, malignity
- Akútní leukemie
- Leukemoidní reakce: vyplavení do krve větší množství granulocytů/prekurzorů
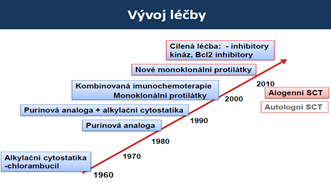
Terapie:
- 1.linie léčby: inhibitor tyrozinové kinázy (TKI)
- 1.generace TKI – imatinib: iniciální dávka 400mg/den (Glivec), terapie je doživotní
- když jsou pacienti rezistentní na imatinib, nebo nereagují na ni podáme 2. generace TKI
- 2.generace: dasatinib (Sprycel), nilotinib (Tasigna), bosutinib (Busilvex)
- 3.generace: ponatinib (Iclusig)
- Jediná kurativní terapie: alogenní transplantace kmeňových buněk (mladší pacienti)
- indikace ak selhá 1.linie léčby TKI léky