úvodní dávka je 500mg/den, postupne můžeme dávku zvyšovat dle tolerance pacienta až na 2500-3000 mg/den
ma dlouhodobý příznivý efekt na kardivaskulární morbiditu a mortalitu
Mechanizmus účinku:
zvyšují senzitivitu jater a svalové tkáně k inzulinu
tlumí glukoneogenezi i vstřebávání glukózy v tenkém střevě
↑inzulinovu senzitivit tým, že ↓jaterní glukoneogenezi a ↓glykemii nalačno)
Kontraindikace:
renální insuficience (clearence kreatininu/eGRF pod 30ml/min, mezi 30-60ml/min metformin taky nedávame, ak už metformin pacient užíva snížime dávky na polovinu)
srdeční selhání
tězké tkáňové hypoxické stavy
těžká respirační insuficience
ak se nerespektuje KI: riziko vzniku závažné ale vzácne komplikace: laktátové acidózy
Nežadoucí účinky: nevolnost, meteorismus, průjem (do několik týdnů ustupuje)
před chirurgickym výkonem: vysadit 48 hodin před
před podaním jodové kontrastní látky: vysadit 48 hodin před
2. Derivaty sulfonylurey: glimepirid, gliklazid
Mechanizmus účinku:
zvyšují sekreci inzulinu z β-buněk
snižují postprandiální (po jídle) glykemii
Nežadoucí účinky:
hypoglykemie (při předávkování)
↑hmotnost (zvyšují chuť k jídlu)
Kontraindikace: renální insuficience
u obézního diabetika je chybou podat derivát sulfonylurey jako lék 1.volby, protože neovlivňují inzulínovou rezistenci a ↑hmotnost
3. Glitazony: thiazolidindiony, pioglitazon
Mechanizmus účinku: aktivují prostřednictvím jaderného receptoru PPAR-γ transkripci genů, odpovídajících za metabolické účinky inzulinu a zvýšení inzulinové senzitivity
léčba je v kombinaci s metforminem/deriváty sulfonylurey
Inkretiny jsou peptidy uvolňované z trávicího traktu, které napomáhají dosažení glukózové homeostázy. Hlavními inkretinovými hormony jsou glukagon-like peptid-1 (GLP-1) a glukózo-dependentní inzulinotropní polypeptid (GIP). GLP-1 stimuluje sekreci inzulinu jako odpověď po příjmu potravy, inhibuje sekreci glukagonu a zpomaluje vyprazdňování žaludku. Vede tím k výraznému snížení výkyvů glykémie po jídle.
Mechanizmus účinku: ↑sekrece inzulinu pouze při hyperglykémií, při normalní hladine cukru v krvi nebo snížene sekrece inzulinu neovplivní) a ↓sekrece glukagonu
v kombinaci s metforminem/sulfonylerey
↓hmotnost: je velmi výhodné u obéznich, příznivě na tlak (snižují tlak)
5. Glifloziny: dapagliflozin
Mechanizmus účinku:
inhibuji transportér v proximálnich tubulech ledvin a zvyšují glykosurii o cca 70g/den → kompenzace diabetu
mírně snižují hmotnost
snižují krevní tlak
Nežádoucí účinky: urogenitální infekce
v kombinaci s ostatnými p.o. antidiabetiky
6. Glinidy: repaglinid a nateglinid
působí rychle, takže jsou ideální k užívání s jídlem ke kompenzaci postprandiální hyperglykémie
TERAPIE INZULINEM
jediná léčba u DM 1.typu, léčba i u řady diabetiku 2.typu
endogénni sekrece u 70kg cca je 40IU/den:
bazální sekrece (neovlivněna příjmem potravy cca polovina)
postprandiální sekrece (vyvolána příjmem potravy, druhá polovina)
u DM 1.typu: nutne pokrýt celou denní dávku: 35-45IU/den
u DM 2.typu: není nutné pokrýt celou denní potřebu (10-15j.)/nebo/ je inzulinova rezistence a tá potřeba je zvýšena = dávka 80 a více j./den
Definice: je chronické endokrinně-metabolické onemocnění v důsledku absolutniho nebo relativniho nedostatku inzulinu, postihující metabolismus sacharidů, lipidů, proteinů a vodní a minerálové hospodaření. Skupina metabolických onemocnění, pro které je společná chronická hyperglykémie.
glykosurie (vzniká ak překročí renálni absorpční prah cca 10-12 mmol/l) ak je výrazná tak vzniká → osmotická polyurie → dehydratace → ↑pocit žízně → polydipsie (↑příjem tekutin-chorobny)
hubnuti → je způsobeno ztrátou tekutin, ↓příjem jídla (nechutenství)
Zrak → změny koncentrace glukózy a osmotických poměru v oku
DM 2.typu: často oligosymptomaticky (jenom celková únava) nebo asymptomaticky průběh → zjištěno náhodně (laboratorní vyšetření)
Prognostický význam stanovení hladiny ADAMTS13 Ag:
↓ ADAMTS13 Ag v akutní fázi onemocnění = ↑ mortalita
↑ hladina ADAMTS13 Ag = klinické zlepšení
Mechanismus: poškození endotelu cévní stěny – uvolnění multimerů vWf. do cirkulace + depozita fibrinu v místě poškození + destičkové agregáty + zúžení cévy – mechanické rozrušení ery – hemolýza
Patofyziologie:fragmentace krvinek – schistocyty – zvýšený zánik ve slezině, při intravaskulární hemolýze + současně aktivace koagulačního systému – DIC s tvorbou mikrotrombů v cirkulaci
1. Při akutní hemolýze s pozitivním Coombsem okamžitě kontaktuj transfuzní odd.:
identifikace protilátky – chladová/tepelná a nejlépe stanovení jejího titru
vysycení séra pacienta od autoprotilátek pro zajištění kompatibilního křížového testu (1-2 pracovní dni, ne víkend)
2. Rozhodnutí zda trasfundovat ERY?
Dle kliniky/rychlosti rozvoje anémie, i malé množství krve může zachránit život
V urgentním případě transfundujeme ERY inkompatibilní v křížového testu z vitální indikace, dle pravidla:
U žen které nebyly těhotné a trasnfundované a mužů kteří nebyli trasnfundovaní máme skoro jistotu že nemají přítomnou allo-protilátku: stačí ABO a RhD kompatibilita
U ostatních v případě urgence vyžaduji podrobnější výběr ERY dle shodnosti fenotypu navíc v C,c, E,e, Kell, Kid a S/s
Vždy biologický in vivo test kompatibility: po premedikaci steroidy, rychle podat 20ml krve a počkat 20 min., zda se nevyskytne rakce…
3. Jaká imunosupresní therapie u život ohrožující hemolýzy?
metylprednisolon ve vysoké dávce (2x 0,5-1,0gr/den)
Plazmaferéza
IVIG – mají v therapii AIHA pouze krátkodobý efekt, ale v případě urgence mají své opodstatnění. (podávat po PLF)
Definice: je pokles počtu destiček. Vzniká nepoměrem mezi novotvorbou a zánikem trombocytů. Příčinou je selhání tvorby destiček v kostní dřeni nebo jejich urychlený zánik.
Klinický obraz: spontánní krvácive projevy až při poklesu pod 30-10×109/l
Trombocytopenie ze snižené tvorby krevních destiček:
Trombocytopenie amegakaryocytární
Trombocytopenie megakaryocytární
Myelodysplastický syndrom
Trombocytopenie ze zvýšeného zániku krevních destiček:
Imunní trombycytopenie
Trombotická trombocytopenická purpura
Hemolyticko – uremický syndrom
Heparinem indukovaná trombocytopenie
Trombocytopenie ze snižené tvorby krevních destiček:
Trombocytopenie amegakaryocytární
Trombocytopenie megakaryocytární
Myelodysplastický syndrom
1.TROMBOCYTOPENIE AMEGAKARYOCYTÁRNÍ
Definice: je trombocytopenie ze snížené tvorby krevních destiček (útlum novotvorby). Množství megakaryocytů v kostní dřeni je snížené, nebo úplně chybějí. Útlum býva izolovaný nebo postihuje celou myeloidní řadu.
Dělení:
Vrozené – vźacné
Získané (sekundární) – po léčbě myelotoxickýmí látkami, ionizující záření, vírové infekce, infiltrace kostní dřeně nádorem (leukemické buňky, metastáza karcinomu), při pokročilé fibróze kostní dřeni při myeloproliferatívím onemocněním, aplastická anemie
Etiopatogeneze: předpokláda se účast (při idiopatických) imunitnních dějů
Diagnóza: vyšetření kostní dřeně
Klinický obraz: spontánní krvácive projevy: až při poklesu pod 30-10×109/l
Terapie:
Sekundární příčiny – terapie primárního onemocnění, nebo odstraněnní vyvolávajícího agens
Část reaguje na imunosupresivní terapii
Může být indikováná alogenní transplantace kostní dřeně
Definice: je trombocytopenie ze snížené tvorby krevních destiček. V kostní dřeni je normálný nebo zvýšený počet megakaryocytů ale vykazují různé tvarové odchylky.
Patří sem:
Trombocytopenie u megaloblastových anémií
Paroxysmální noční hemoglobinurie
Myelodysplastický syndrom
Trombocytopenie ze zvýšeného zániku krevních destiček:
Imunní trombycytopenie
Trombotická trombocytopenická purpura
Hemolyticko-uremický syndrom
Heparinem indukovaná trombocytopenie
TROMBOTICKÁ TROMBOCYTOPENICKÁ PURPURA
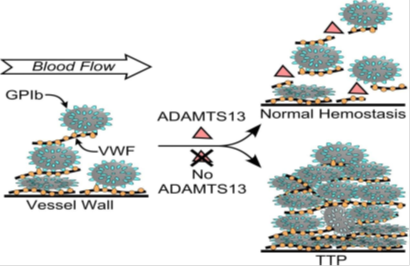
Definice: je trombocytopenie ze zvýšeného zániku trombocytů s klasickým klinickým obrazem zahrnujícím pentádu: hemolytickou anémii, trombocytopenii s krvácivými projevy, s neurologickou symptomatológií a centrálními febriliemi a ďalším orgánovým postižením hlavně ledviny. Je často poddiagnostikovaná, bez terapie umírá 90% pacientů.
Epidemiológie: 3 – 7/1 milion/rok, průměrný věk při záchytu 35 let
Etiopatogeneze:
Etiologie: deficit metaloproteinázy: ADAMTS13, v důsledku autoprotilátek
Patogeneze: v normálních hemostatických poměrech multimery vWF adherují na endotel cévní stěny a trombocyty adherující na vWF svým membránovým proteinem GPIb. Aby nedocházelo k dalšímu narůstání trombu jsou multimery vWF štěpeny ADAMTS13. V případě deficitu ADAMTS13, pokračuje akumulace trombocytů, dochází k trombotizaci v mikrocirkulaci, ischemizaci tkání a TTP.
Dělení:
Familiární: raritní
Upshaw-schulman syndrom vznikající mutací ADAMTS13 g.
trombocytopenie (většinou < 40 tisíc, průměr 16 tisíc)
+/- renální postižení
+/- neurologická symptomatologie e) Možno vyloučit jinou příčinu trombotické mikroangiopathie (TMA)
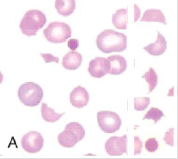
a) mikroangiopatická hemolytická anémie:
Negativní Coombsův test
↑ bilirubin
↑ LDH
↓ haptoglobin
↑ počet schistocytů
(běžná laborantka i o službě v nátěru odečte zvýšený počet schistocytů a zároveň vyloučí shluky trombocytů při EDTA – pseudotrombocytopenii)
Coombsův test (antiglobulinový test, AGT) slouží k testování přítomnosti protilátek proti erytrocytům. Rozlišujeme 2 varianty Coombsova testu:
Coombsův test přímý slouží k diagnostice hemolytických anémií způsobených antierytrocytárními protilátkami. Pomocí protilátek proti antierytrocytárním protilátkám testujeme přítomnost těchto antierytrocytárních protilátek na membráně erytrocytu.
Coombsův test nepřímý slouží k detekci antierytrocytárních protilátek v krevní plazmě u pacientů po opakovaných krevních transfúzích. Pomocí protilátek proti antierytrocytárním protilátkám testujeme přítomnost antierytrocytárních protilátek v krevní plazmě.
Diferenciální diagnóza:
Maligní hypertenze – může způsobit mikroangiopatickou hemolytickou anémii, trombocytopenii, renální selhání i neurologické symptomy.
Systémová infekce – septikémie jakékoliv etiologie (bakterie, plísně, viry) může vyvolat trombocytopenii a mikroangiopathickou hemolytickou anémii. Dif.dg. horečky, zimnice, plicní infiltráty nejsou u TTP časté.
Systémová malignita – může způsobit trombocytopenii i anémii. Dif.dg. postižení jater nebo plic je raritní u TTP. Početní změny v leukocytech (nahoru/dolu) ozřejmí mikroskopický diferenciál z nátěru event. punkce KD.
Preeklampsie/HELLP syndrom – může vyvolat trombocytopenii, mikroangiopathickou hemolytickou anémii, renální selhání i mírné neurologické příznaky.
Terapie:
1. Výměnná plazmaferéza (PLF)
dochází k substituci ADAMTS13 a zároveň odstranění autoprotilátek inhibujících její aktivitu
1x denně, 1-1,5x plazmatického objemu pacienta
náhradním roztokem je čerstvě zmražená plazma
2. Čerstvě zmražená plazma (pouze) pokud není PLF dostupná ihned, je akceptovatelné její podání jako bridge k PLF
v dávce 30-50 ml/kg/den.
3. Transfuze trombocytů:
dogma, že substituce trombocyty je absolutně kontraindikována, již neplatí
při závažném krvácení se trombocyty substituují
při přípravě na invazivní výkon (například dialyzační kanyla) trombocyty profylakticky nepodávám, ale jsou v rezervě na TO k okamžitému podání v případě komplikací
profylakticky trombocyty nepodám i při velmi nízké hodnotě
4. Kortikosteroidy (inhibice tvorby autoprotilátek proti ADAMTS13)
Nejednoznačná doporučení dávkování, ale pacienti v dobré kondici bez neurologických symptomů:
Prednison 1mg/kg/den p.os.
Pacienti akutně nemocní:
Methylprednisolon 2mg/kg/den iv., ve 2 dávkách
U pacientů s podezřením/potvrzení na E.Coli enteritidu/infekci steroidy neindikovány.
V praxi – zahájíme léčbu jako idiopatickou TTP a po několika dnech a určení dg. E.Coli TTP-HUS se kortikoidy detrahují
Hodnocení léčebné odpovědi:
Dle hodnot trombocytů:
vzestup očekáváme 2 – 3 den PLF
normalizace dosáhneme za cca 1 týden
společně se vzestupem Trombocytů, klesá bilirubin, LDH a schistocyty
Zlepšení neurologických symptomů může být i prvním příznakem terapeutické odpovědi, plná reverzibilita neurologického deficitu je pravděpodobná
Korekce anémie je významně pomalejší než u trombocytů
Korekce renálního selhání také pomalejší, úplná recovery není jistá
Postup po dosažení remise:
Pokud jsou 2 dny stabilní trombocyty >150 tisíc:
prodloužím interval PLF na obden, cca na 1 týden
redukce steroidu
Pokud je počet trombocytů stabilní cca 1 týden od zahájení redukce léčby:
pacient je k překladu na standardní oddělení
zde ukončujeme PLF, CŽK EX
převádím na steroidy p.os., jejich další redukce pomalejší á 1 týden
Refrakterní k 1. linii
po 4 – 7 dnech plné terapie bez vzestupu trombocytů
po přechodném zvýšení Trombocytů opět dochází k poklesu často s novými neurologickými symptomy
Intenzifikace terapie:
Metylprednisolon 1000mg/den na 3 dny
Výměnná PLF 2xdenně
Rituximab 375mg/qm 1xtýdně, 4 dávky
Další možnosti:
Imunoabsorbce na Stafylokokových A kolonách
Posílení imunosuprese: CFA, vincristine, CSA
Prognóza:
Závažnost správné diagnostiky a terapie TTP vyplývá ze skutečnosti, že mortaliza neléčených pacientů je 90%
V současnosti docilujeme ČR v 75 – 85 % léčených případů
Celkově dochází k cca 35% k relapsu v 5ti letech s nejvyšším rizikem relapsu 1. rok po ukončení terapie.
Polékové, těhotenské TTP většinou nerelabují
HEPARINEM INDUKOVANÁ TROMBOCYTOPENIE
(HIT)
Heparinem indukovaná trombocytopenie I. typu:
pokles trombocytů, nikdy pod 100×109/l
neimuními mechanismy, zcela benigní průběh, sama odezní
10 – 30 % pacientů dostávajících heparin
většinou 4 den podávání heparinu
nevyžaduje léčbu, heparin se neukončuje
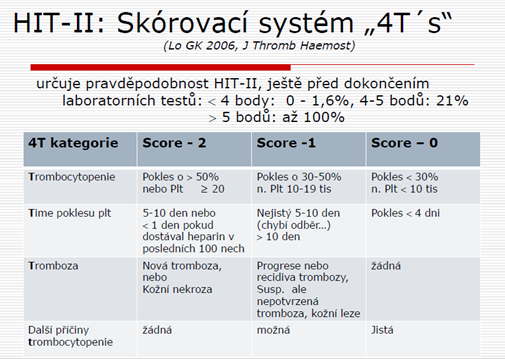
Heparinem indukovaná trombocytopenie II. typu: je klinicko – patologický syndrom definovaný:
trombocytopenií
+/- trombozou
v jasné časové souvislosti s podáním heparinu
způsobený přítomností HIT protilátek
Etiopatogeneze:
HIT II. typu: je imunního původu
Heparin se váže na destičkový PF4 = deštičkový faktor 4 → komplex PF4/heparin může být rozeznán jako cizorodý antigen a dojde ke tvorbě autoprotilátek: anti-PF4 /heparin complex IgG (HIT-Ab)
HIT – protilátky způsobí:
trombocytopenii
další aktivací trombocytů a monocytů se zvýšenou produkcí tkáňového faktoru, je paradoxně při současné trombocytopenii významně zvýšená incidence trombózy
Epidemiologie:
Vytvoření a detekce HIT – protilátek:
U pacientů dostávajících heparin není podmínkou rozvoje klinické HIT:
kardiologický pacient: 2 – 5 % HIT – Ab
ortopedická operace: 15 – 30 %
kardiochirurgické výkony: 30 – 70 %
Incidence klinické HIT je výrazně nižší:
UFH (nefrakciovaný heparin): 1 – 5 %
LMWH (nizkomolekulární hepariny): 1 %
Kardiochirurgie: 2,4 %
ICU celkově: 2%
Dávkování heparinu:
i profylaktické dávkování zvyšuje riziko tvorby HIT – Ab
klinická manifestace HIT je častější u heparinu v terapeutickém dávkování
Diagnóza:
1. Trombocytopenie:
První projev HIT (85% pacientů): je pokles trombocytů pod 150 tisíc nebo o 50% vstupní hodnoty → typicky poklesnou na hodnoty 40 – 80tisíc → pouze u 5 – 10 % pacientů poklesnou pod < 20 tisíc
Zvláště u těžké trombocytopenie musíme pátrat po dalších možných příčinách!
2. Doba vzniku trombocytopenie:
Typický průběh: 5 – 10 dní po zahájení heparinu
„rapid onset“ HIT: v průběhu 24 hodin od zahájení heparinu, v důsledku již přítomných HIT – Ab u pacientů, kteří dostávali heparinu v posledních 100 dnech. Možné alergické projevy zimnice, třesavka, horečka
3. Trombotické komplikace:
u 30 – 70 % HIT jsou přítomné trombotické komplikace
!!! v případě vzniku trombózy po zahájení plné heparinizace, nelze vyloučit HIT, i když nedošlo k poklesu trombocytů
až 40% trombóz vzniká 3 dni před vznikem trombocytopenie
4. Další komplikace:
i při poklesu trombocytů < 20 tisíc, riziko závažného krvácení je nízké
u 10 – 20 % pacientů se průměrně 8. den po nasazení heparinu vytváření kožní léze, od erytému po nekrózu
5. Laboratorní diagnóza: ELISA IgG – HIT – Ab
Negativní – vylučuje HIT
Positivní (viz. výše) – nepotvrzuje diagnózu, nutno v kontextu klinické manifestace a laboratorních nálezů
Ukončit léčbu heparinem, vyloučit přítomnost heparinových zátek, heparin-coated katétrů nebo filtrů
Pokud není závažné krvácení, profylaktické podání trombokoncentrátů je nevhodné, zvyšuje riziko trombotických komplikací a vzestup počtu trombocytů je mizivý
Použití antagonistů vitaminu K je kontraindikované – výrazné zvýšení rizika trombózy (rychlý pokles proteinu C, versus delší poločas ff.II,VII,IX,X)
2. Zahájení alternativní antikoagulační terapie:
Přímé inhibitory trombinu:
Lepirudin (REFLUDAN 20mg, i.v., Avensis)
Monitorace účinku: 1,5 – 2,0 x norma aPTT, nástup účinku do 15 minut, poločas 1 – 2 hodiny, neni antidotum
Definice: trombofilie (synonymum hyperkoagulační stav) znamená zvýšenou dispozici k tvorbě trombů v cévním systémů, která předchází vlastnímu procesu trombogeneze.
Epidemiologie: výskyt se zvyšuje s věkem >60let
Etiopatogeneze:
Etiologie: multifaktoriální jehož riziko se zvyšuje přítomností různých faktorů: podnět + trombofilie = trombus
Celkově: slabost, svědění kůže (po koupeli), pletora kůže, vyšší TK, splenomegalie (70%), hepatosplenomegalie (40%)
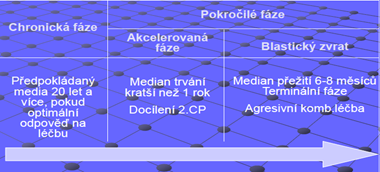
Průběh nemoci: 3 fáze
Pre-polycytemická fáze: mírna erytrocytóza
Rozvinutá polycytemická fáze: signifikantní zmnožení krevní masy
Post-polycytemická fáze: rozvoj myelofibrózy, pancytopenie s přítomnosti mladších forem granulocytární a erytroidní řady v krvi, výrazná splenomegalie, extramedulární hematopoéza
Terminální stadium: může přejít do akútní leukemie
Diagnóza:
Anamnéza a fyzikální vyšetření
Laboratorní vyšetření:
Krevní obraz:
↑Hb ( > 185g/l u mužů, > 165g/l ženy)
leukocytóza ( >10 – 12×109/l)
normální /↑ trombocyty
zvýšen erytrocitární a celkový volum
↓EPO
Kostní dřeň: hypercelulární s proliferací všech tří řad, přechod do fibrózy
Průkaz JAK2
trepanobiopsie
Zobrazovací metody:
USG: splenomegalie
Diferenciální diagnóza:
Nepravé polycytemie: ztráta tekutin, ↑TK, diuretika, choroby ledvin, alkohol
Sekundární polycytemie: fiziologické reakce erytropoézy na hypoxii, nadproduke EPO
Hypoxie: choroby plic, cyanotické srdeční vady, arterio-venózní zkraty, methemoglobinémie, karboxyhemogloobinemie, ve vysokých výškách
Terapie:
Cíl léčby– ovlivnit rizika trombotických a krvácivých komplikací a symptomů
První linie léčby: venepunkce/nebo/erytrocytaferéza a antiagregační terapie ASA (kyselina acetylsalicylová) 50 – 100 mg/den, trvalá při anamnéze plicní embolie nebo hluboké žilní trombózy
Cytoredukční léčba: interferon alfa (INF – α), ev. hydroxyurea u starších pacientů (> 65 let), při výrazné trombocytémii ev. přidat anagrelid
Druhá linie léčby: ruxolitinib (selektívní inhibitor JAK1+2 kináz)
Alogenní transplantace: kurativní možnost léčby (s rizikem, proto až ve stadiu počínající postpolycytemické myelofibrózy)
Definice: je chronické myeloproliferatívní onemocnění postihující primárně megakaryocytární řadu charaktizováno trombocytózou ( > 450×109/l) se zvýšeným počtem megakaryocytů v kostní dřeni a se zvýšeným sklonem k trombotickým či krvácivým projevům.
Epidemiologie: 0,6 – 2,5/100.000/rok
Etiopatogeneze:
Etiologie: neznáma, 50% mutace JAK2 genu
Klinický obraz:
30% pacientů bez příznaků
krvácivé projevy: epistaxe, krvácení do GIT
trombotické projevy: okluze postihuje periferní řečiště s ischemickými atakami v akrech (prsty rukou/nohou), parestézie, vzácně rozvoj gangrény
Mikrovaskulární symptomy: přechodné poruchy vizu, závratě, bolesti hlavy
může vzácně přejít do myelofibrózy ale NIKDY do akutní leukemie
Diagnóza:
Anamnéza a fyzikální vyšetření:
slezina: 1/3 mírně zvětšena
Laboratorní vyšetření:
Krevní obraz:
trombocytóza (>450×109/l)
leukocytóza mírná (15×109/l)
Hb (hemoglobin) v normě
leukoerytroblasty nejsou přítomny
Vyšetření JAK2 (odlíšení od reaktívní trombocytózy)
WHO dg. kriteria: Nutno splnit všechna 4 kriteria
trombocyty trvale ≥ 450×109/l
histopatologický obraz KD
nesplnění kriterií PV, MF, CML, MDS
přítomnost JAK2 V617F nebo jiného klon.markeru.
Diferenciální diagnóza: reaktívní tromcotytemie (chronické záněty, malignity polytraumata, při krvácení, po operaci)
Terapie:
Hlavní cíl – snížit rizika trombotických a krvácivých komplikací a mikrovaskulárních symptomů
Rychlý efekt má trombocytaferéza
Rizikové faktory pro vznik trombózy: věk, počet trombocytů, přítomnost dalších trombofilních faktorů, kouření, obezita, hormonální antikoncepce; mutace JAK2 genu
Při venózní TEN – celoživotní antikoagulační terapie
Trombocyty: do 1000×109/l: kyselina acetylsalicylová či IFN – α
Trombocyty: > 1500×109/l: hydroxyurea či opakovaně trombocytoferéza
Prognóza přežití – většinou nekrátí život pacienta, léčba celoživotní
Definice: je klonální myeloproliferatívní onemocnění s postupnou náhradou krvetvorby v kostní dřeni fibrózní tkání a proliferací krvetvorné tkáně v extramedulární lokalizaci.
bez klinických příznaků nebo neurčité příznaky: slabost, hubnutí, noční poty, únava, periferní edémy
Fibroticka fáze:
výrazná fibróza až osteoskleróza kostní dřene, leuko-erytroblastóza v periferní krvi
projevy cytopenie, hepatosplenomegalie, trombembolie, portální hypertenze, ascites, splenomegalie často až do malé pánve (časté infarkty), pocity plnosti břicha, plynatost, poruchy pasáže, dušnost
Při progresi: teploty, kachektizace, bolesti v levém podžebří ze splenomegalie s extramedulární hematopoezou, infekční komplikace, selhání hematopoezy, u 20% nemocných rozvoj akutní leukemie
nejsou vyplaveny mladší formy erytroidní a granulocytární řady
Fibrotická fáze:
Anamnéza a fyzikální vyšetření:
výrazná splenomegalie a hepatomegalie (portální hypertenze a ascites)
Laboratorní vyšetření:
Krevní obraz:
pancytopenie s leukoerytroblastickou reakcí a přítomnosti slzičkovitých ERY
Z přednášky:
Diagnóza: splnění 3 hlavních a 2 vedlejších kritérií
Hlavní kriteria:
Atypická hyperplazie megakaryocytů a retikulinová nebo kolagenní fibróza nebo atypie megakaryocytů a hypercelularita KD s myelodiní metaplazií a erytroidní hypoplazií
JAK2V617F nebo jiný klonální marker, není-li pak vyloučit sekundární MF (nádorové onemocnění)
První příznaky: únava, bolest kloubů, svalů, subfebrilie, pocit tlaku pod levým obloukem žeberním (splenomegalie), bolest (mechanické zvětšovaním, napínaní pouzdra), noční pocení, nechutenství, hubnutí, hepatomegalie…
Akcelerovaná fáze: nádorové kmeňové buňky se stávají agresivnějšími a zvyšují rychlost proliferace, ztráci citlivost na léčbu